


New tools inspire hope for understanding neurological disease

To fix something built wrong, learn how it works when it’s built right. Following this logic, Cornell scientists have shed new light on an essential life process by designing novel functional tools for addressing neurological disease. Published in the EMBO Journal in February 2013, Their study gives insight into how cells build internal transportation routes— discoveries that could unlock treatments for two neurodegenerative diseases, Hereditary Spastic Paraplegia (HSP) and Hereditary Sensory Neuropathy (HSN).
The study found how the ¬¬protein Atlastin-1 morphs to connect thin, pipe-like membrane tubules within a cellular compartment known as the endoplasmic reticulum (ER). Atlastin connects the ER’s transportation network, but defective forms of Atlastin-1 have altered functions that can lead to degradation of spinal neurons, resulting in HSP or HSN.
This group of genetic neurodegenerative disorders can cause weakness, pain, spasms, muscle control loss, blindness, mental retardation, dementia, and epilepsy. Most cases go undiagnosed, because only five of the 30-plus HSP subtypes show up in genetic testing. For one such type, the study opens new non-genetic ways to test how mutations in Atlastin disrupt its function and lead to disease based on Atlastin’s three-dimensional structure.

“We’ve found the sequence of shapes and actions Atlastin takes to merge the ER’s pipelines,” said Dr. Holger Sondermann, structural biologist at Cornell’s College of Veterinary Medicine, whose group conducted the study. “We know of nearly 40 different genetic mutations in Atlastin linking to HSP and HSN. Now that we know how Atlastin works physically, we have a new diagnostic tool to characterize mutants that doesn’t rely on genetics. This enables us to look at how mutants differ, hopefully getting to the root of these diseases and finding ways to stop or manage them.”
Using a soluble fragment of Atlastin, Laura Byrnes, a graduate student in Sondermann’s group, grew protein crystals and exposed them to X-rays at the Cornell High Energy Synchrotron Source. By analyzing how X-rays reflected off the crystals, the team determined three distinct molecular structures Atlastin adopts as it works.
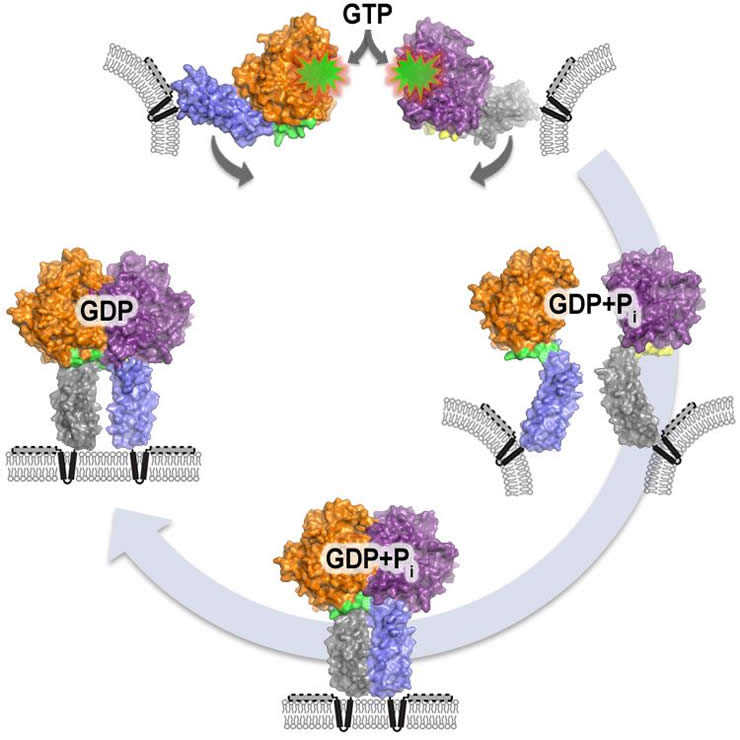
Next, with an advanced technique called fluorescence resonance energy transfer (FRET), Sondermann’s group developed a test to move from these snapshot portraits to the molecular equivalent of real-time video, monitoring Atlastin live to see the order in which it can take on different shapes while performing its tasks.
They found that pairs of Atlastin proteins work in synch, changing through structural states to come up with the energy required to draw them close enough to fuse their ER membrane tubes together. Two Atlastins temporarily combine into one super molecule, a process called dimerization, and work together to connect their respective pipes. This process is controlled by nucleotides, small energy-carrying packets that bind to Atlastin.
“The sequence of events we uncovered shows how Atlastin uses the energy stored in the nucleotide guanosine triphosphate to time the shape changes and eventually dimerize,” said Sondermann. “The type of nucleotide Atlastin binds determines whether it can go through a functional cycle or dimerize. Tinkering with this crucial ability is likely to contribute to disease. Using the tools developed in this study will lead to a better understanding of how Atlastin works and where mutants fail, which may open doors to better testing and future treatments for HSP.”


